Introducing Open Chemistry at Kitware

I have been working on open-source chemistry applications since the summer of 2007, when I took part in the Google Summer of Code program as a student at the end of my Ph.D. Upon completing my Ph.D. at the University of Sheffield, UK, I moved to the University of Pittsburgh as a postdoctoral researcher in the newly-formed Hutchison [1] group where I continued to work on open-source chemistry tools and applications with Professor Geoffrey Hutchison. Most of the development I did there was to further our research goals, and went into the open-source projects Avogadro [2] and Open Babel [3].
In 2009, I joined Kitware and continued some of these collaborations in my spare time. I heard in late 2010 that my Phase I SBIR “Open, Cross Platform Chemistry Application” [2] had been accepted under topic A10-110 by the Engineering and Research Development Center (ERDC). I was very excited to be able to continue my work in open chemistry, and move Kitware into a new area of development looking at chemical and biological systems. The proposal was to extend Avogadro, develop new applications for storage and retrieval of chemical data, integrate high performance computing (HPC) resources on the desktop, and more tightly integrate the GAMESS [4] code into Avogadro.
This article will go over some of the highlights of the development work in the Phase I project, and then discuss the work we intend to do in Phase II of the SBIR. This work is largely taking place at Kitware, but there are key collaborations formed as part of the Blue Obelisk movement [5], the Chemical Structure Resolver from the National Institutes of Health (NIH) [6], and the open Quixote collaboration [7] to standardize data in computational quantum chemistry.
The Typical Workflow
The modern computational chemist has access to massive amounts of data, and powerful codes for performing full electronic structure calculations of a given chemical structure, or exploring the configurations of a system as it evolves over time or in the presence of external stimuli. A void which I saw during my Ph.D. research, and several of my collaborators saw, was an open tool that integrates the best of these codes in a workflow that has a lower learning curve so that new researchers can get up and running more quickly.
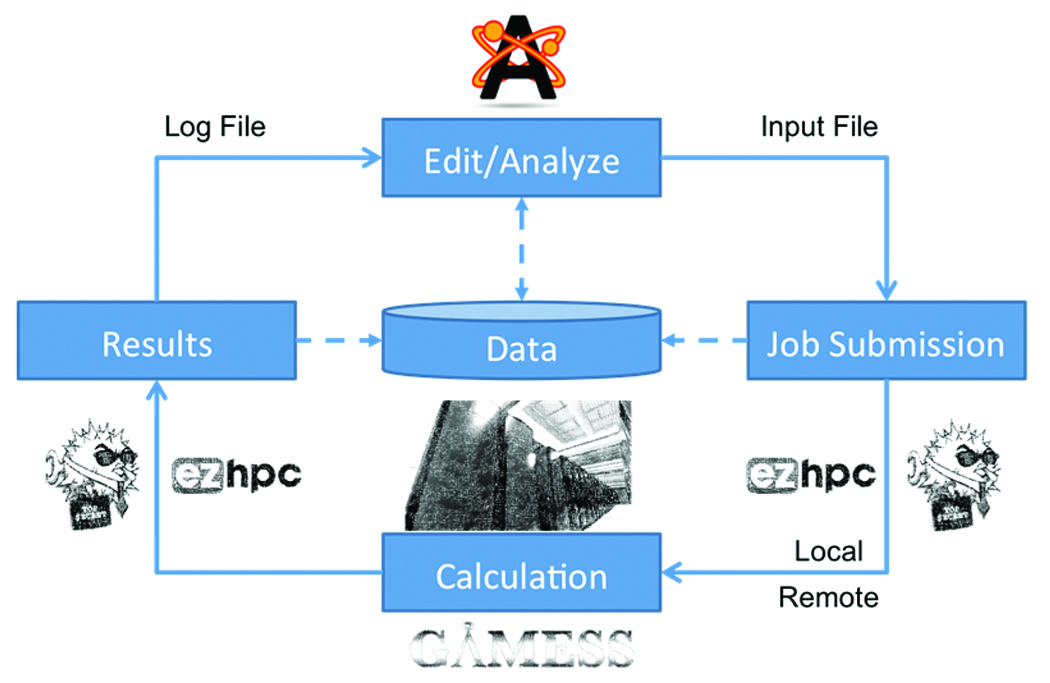
Figure 1. Typical workflow of a computational chemist at present.
When a new student joins a research group, they typically need to obtain a license for a chemical editor before they can start producing results. This often only works on Windows, requiring students to learn how to use the editor in order to draw structures and save them in an appropriate format. They often need to search for a good starting structure (this might be drawn in an editor, or downloaded from a database). Once they have a structure with the desired functional groups in the conformation they would like to investigate, they must produce an input file for their tool of choice. This involves saving out the coordinates and types of atoms, and then editing a text file that contains the parameters for the calculation to be performed. There are often several phases of the calculation: initial geometry optimization using a low level of theory, possibly a second geometry optimization, and then some energy calculations or other work.
Most research groups make use of HPC resources, which are typically Linux-based clusters running a queuing system of some type. This means students need to learn how to use SCP to copy their input file(s) to the cluster; SSH to run the code on the cluster; the syntax to submit jobs to the queuing system; how to monitor the queue to check the progress of their job; and finally how to retrieve their results file(s) once the job is complete. It is not normally possible to perform all calculations/research using a single code, and so it is also necessary to learn several input file formats for each of the codes you would like to use. Tools such as Open Babel are often used to translate between the multitudes of chemical file formats in common use today.
Our vision is to lower the barrier to entry, while providing access to the low-level tools for advanced users. As research in this area is so varied, we did not feel it was enough to create a solution for just one research problem, but to create a framework of tools that uses plugins where appropriate to extend these applications at runtime. This will enable students, researchers and faculty to create the system they need for their research problem, and will support plugins needed for specialized research areas. Kitware is uniquely positioned to collaborate with users of these tools and act as an enabler for their research by creating collaborative innovation platforms for the research community.
Vision
Our vision is to create the leading computational chemistry workbench, making premier computational chemistry codes and databases easily accessible to practitioners in the field of chemistry and materials research. This will be accomplished by creating an open, extensible application framework that puts computational tools, data, and domain specific knowledge at the fingertips of chemists. A data-centric approach to chemistry, storing all data in a searchable database, will empower users to efficiently collaborate, innovate, and push the boundaries of what is possible in research.
Design of the Components
During Phase I of the project, we developed several loosely-coupled components to address each area. The five key areas addressed were:
- Databases: storage and retrieval of chemical data
- Input structure editing and input file preparation
- Job submission and retrieval
- Output ingestion, visualization and analysis
- Component integration into a prototype application
This led to the development of several prototype applications and libraries that were demonstrated at the end of the project. Each component had a tight focus on one or two problem areas, but was designed to work as part of a larger workflow to address the needs of computational chemists.
MoleQueue
A prototype system tray resident Qt application named “MoleQueue” was developed. It takes input files from Avogadro over a local socket connection and manages the lifetime of both local and remotely queued jobs. When managing local jobs it creates a simple queue where one job is run at a time; a sandbox directory is used and then the process executed. When the process exits, the job is marked as complete and the user can open the results file in Avogadro.
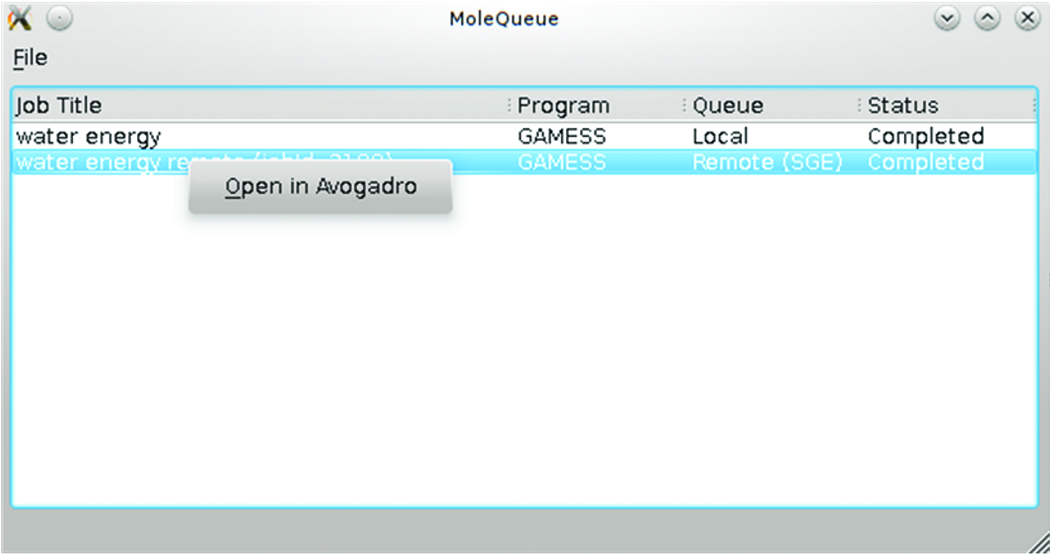
Figure 2. MoleQueue in action with a remote and local GAMESS job.
There is also a remote queue manager, which currently works with the Sun Grid Engine queuing system. The management process is a little more complex as the input file(s) must be moved to a remote staging area. The queuing system command needs to be invoked with the correct arguments to add the job to the queue, and then must be monitored until the job is marked as complete. Once a job is complete, the results files are copied back to the user’s system for analysis and storage. A screenshot of the interface in the application is shown in Figure 2.
ChemData
A prototype chemical data tool, tentatively named “ChemData,” was developed to handle the storage and retrieval of chemical data in the workflow shown in Figure 1. It also performs some 2D cheminformatics visualization of groups of compounds and structural properties. ChemData was developed using Qt for the GUI, MongoDB [8] for data storage and retrieval, and VTK for the 2D visualization. It executes a custom Open Babel program to generate chemical descriptors such as SMILES, InChI and molecular weights to enable searching.
Context menus are provided to open the data files in Avogadro, or to fetch additional information such as 3D structure, 2D chemical representations, or IUPAC names from the NIH Chemical Structure Resolver service. The 2D scatter plot and parallel coordinate representations allow users to select the chemical space they are most interested in, and view that subset of the data in the bottom scatter plot. Tool tips show the chemical abstract service (CAS) identifier and observed/predicted property values for the point the mouse is hovering over. The user can also click individual points and see that point in the table view on the right. From there, they can use the context menu to request more actions, e.g. opening the data file in Avogadro. A typical session with the plots and data table is shown in Figure 3.
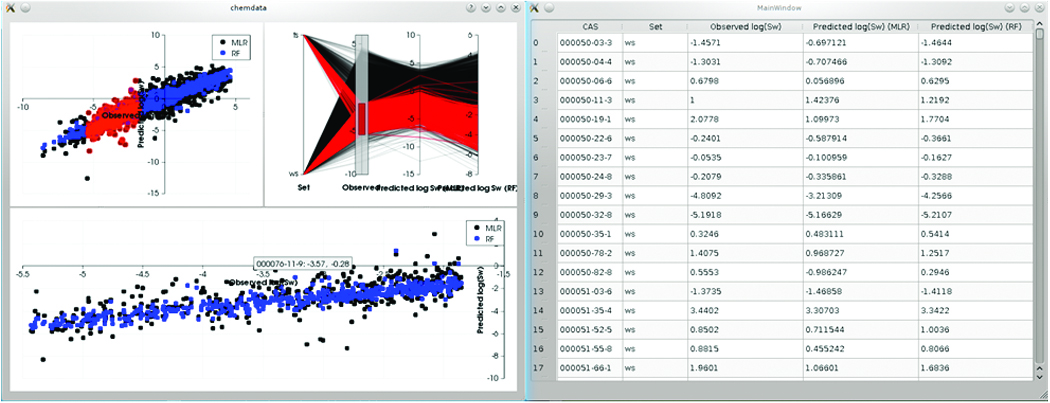
Figure 3. ChemData showing the 2D visualizations (left) and the data table (right).
OpenQube
A library called “OpenQube” was developed largely based on work I did previously in an Avogadro plugin. This library focuses on reading the output from quantum codes, such as GAMESS, and calculating the molecular orbital and electron density cubes, which can be further processed in Avogadro and other applications. At this stage, the OpenQube library is called from an Avogadro plugin and a small test command line program that can be used to drive some of the calculations. The intent is to offload some of this CPU-intensive work to clusters, where available, using a client-server approach to analysis, while keeping the option of using the same code to perform these calculations on the client machine. A typical isosurface of a molecular orbital is shown in Figure 4; the cube was calculated from the output of a quantum code, marching cubes were used to triangulate the surface, and POV-Ray rendered the final image using input generated by an Avogadro plugin.
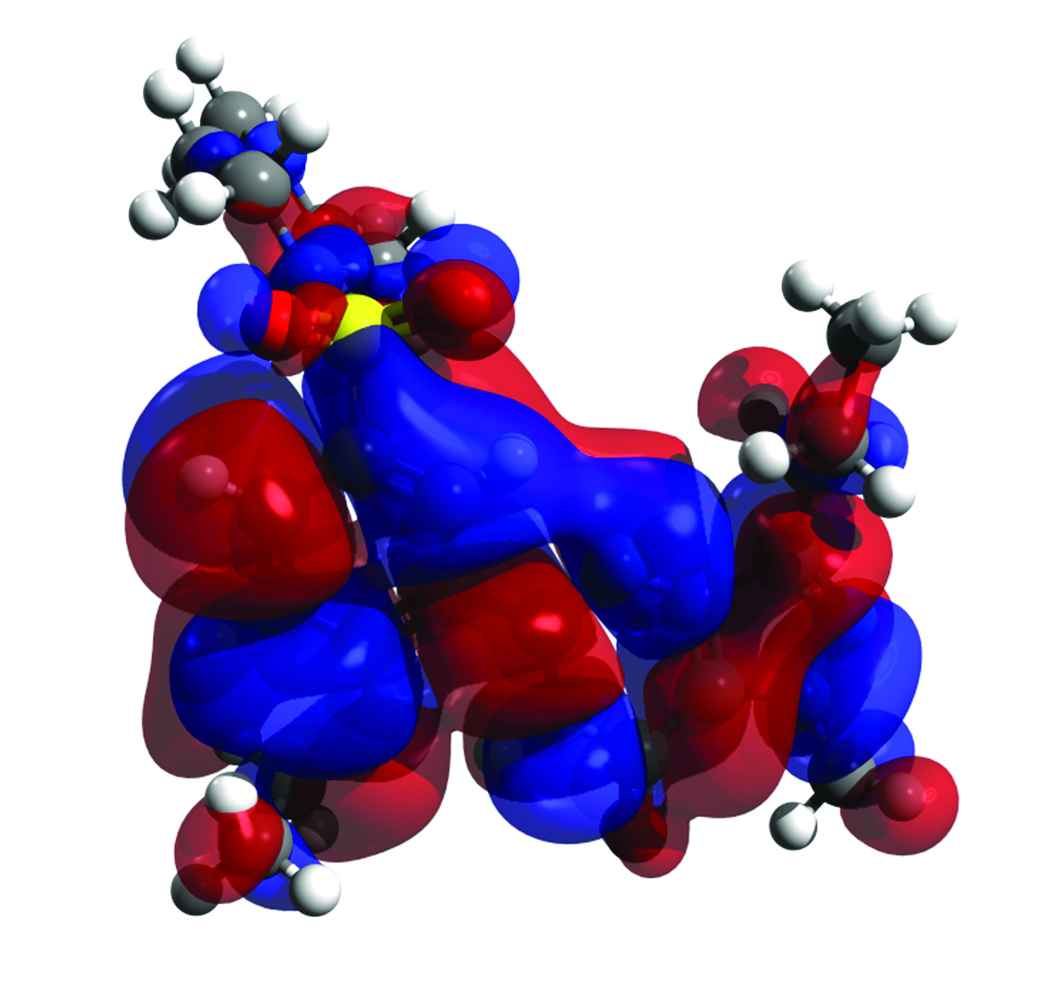
Figure 4. Highest Occupied Molecular Orbital (HOMO) calculated using
OpenQube, generated by Avogadro, and rendered using POV-Ray.
Advanced Visualization in an Avogadro Plugin Using VTK
One of the things that excited me about joining Kitware was the possibility of bringing advanced visualization techniques present in VTK to Avogadro. I developed a small plugin that took the cube data calculated by OpenQube using the electronic structure Avogadro plugin, and used the GPU accelerated volume rendering code present in VTK to examine the electronic structure of the output from GAMESS calculations. It proved quite easy to do this and then add the basic molecular structure rendering to the VTK render window. The results were impressive both in clarity and interactive rendering speeds – even when pushing the dataset size to very fine cubes that took hours to calculate on a 16-core machine. A few examples are shown in Figure 5.

Figure 5: Molecular orbital (left) and electron density (right) rendered by the VTK plugin for Avogadro.
Input Structure Editing and Input File Generation
Most of this work took place inside Avogadro, extending some existing plugins and creating a few new features. The same NIH Structure Resolver Service can be used to fetch a chemical by name, such as “caffeine”. This structure may then be edited before being used as the basis for an input file. The input file dialog was extended to use MoleQueue to run local or remote jobs, without ever touching a terminal. The output file can be opened in Avogadro straight from the MoleQueue dialog, be edited, and then be used to generate new input files before storing them using ChemData.
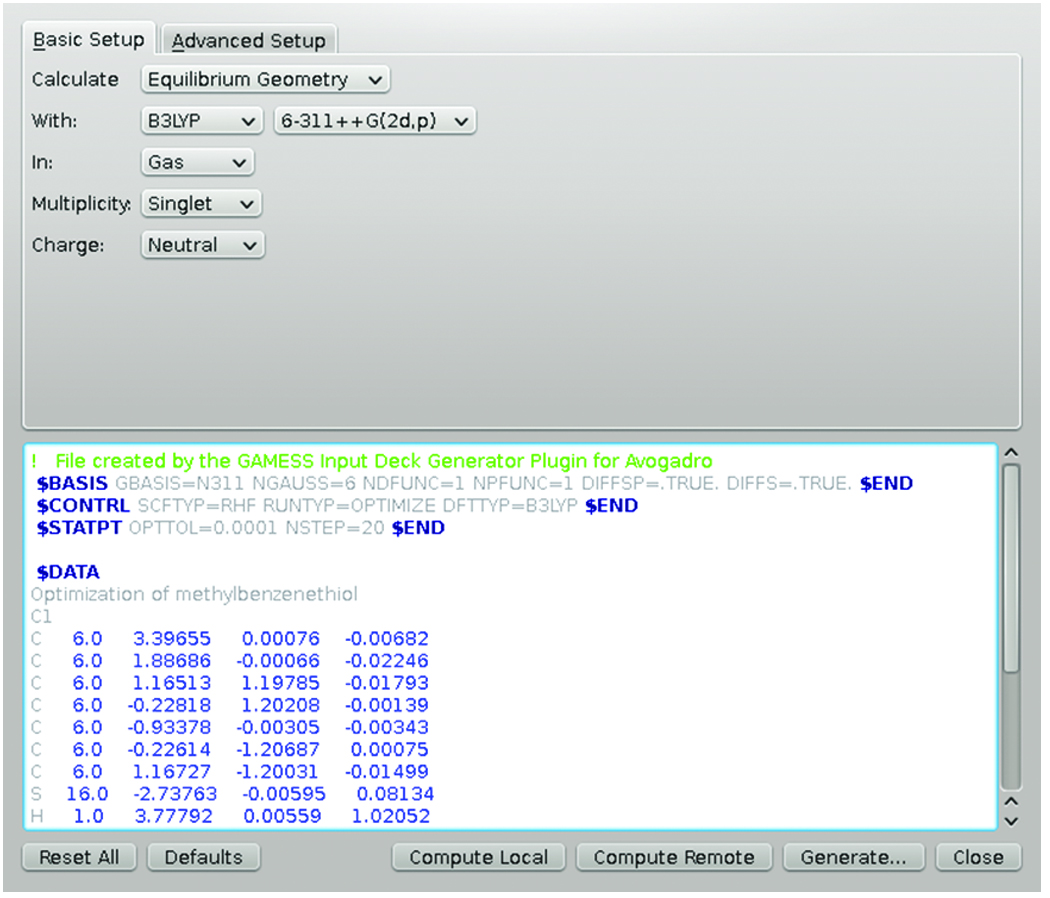
Figure 6: The GAMESS input file dialog in Avogadro.
Figure 6 shows a typical input file dialog in Avogadro for GAMESS. This contains many of the job parameters a user might want to change in the dialog; when these options are changed, they are reflected in the text of the input file immediately. The plugin also features syntax highlighting, enabling advanced users to edit the file directly and receive immediate feedback on any typos or mistakes in syntax. The buttons at the bottom are enabled if the plugin can find a running MoleQueue on the system and the input file, as shown in the dialog, will be sent to MoleQueue when the button is pressed.
Improved Workflows in Chemistry
All of these components were developed and enhanced during the Phase I SBIR, and are poised to revolutionize the workflow of modern computational chemists. Some of the technology, such as MoleQueue, was developed to be useful in applications outside of chemistry. We are already looking into other areas where this would be a useful addition to enable desktop integration of HPC resources. Figure 7 shows the improved workflow, and where these new components fit in with data flowing seamlessly between components.
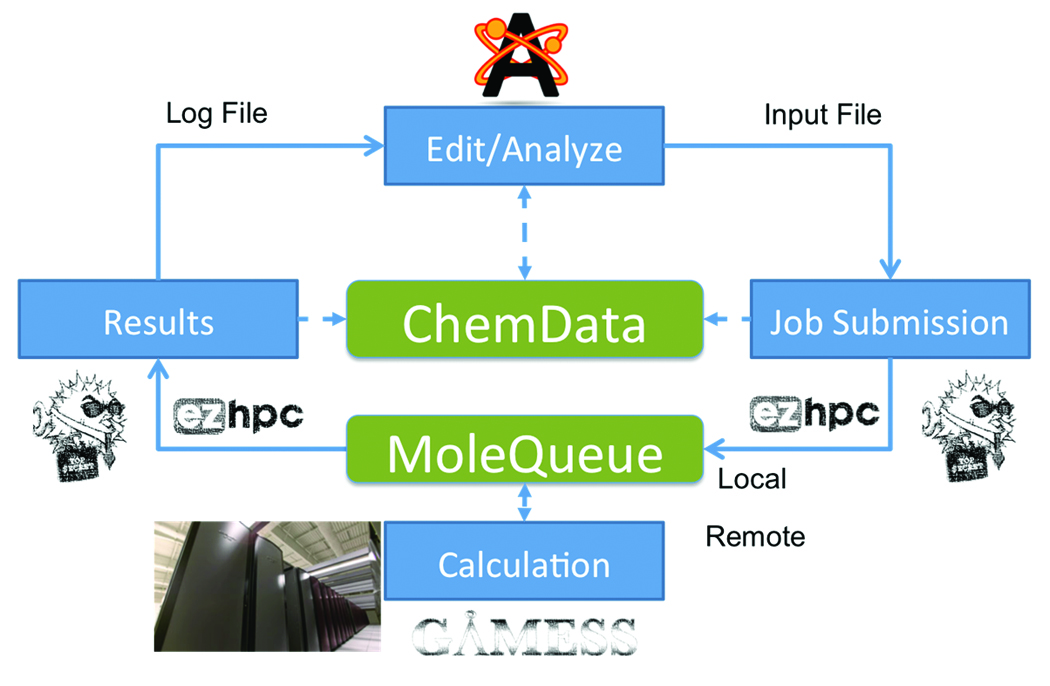
Figure 7: The improved workflow using the open chemistry workbench.
We recently received a notice of award for Phase II of this SBIR project, and I am very excited to push ahead. We will be addressing six major areas in this work:
- Chemical structure manipulation
- Interaction with databases
- Generating input files for chemistry codes, such as GAMESS and NWChem
- Submitting jobs to remote systems, monitoring and retrieving results
- Ingesting and post-processing output from chemistry codes
- Visualization of chemical structure, QSAR, QSPR and other cheminformatics data
There is still a lot to do for Phase II, and we hope to make MoleQueue and ChemData publicly available soon in order to get more feedback. Many of these components are already publicly available and we intend to develop them all in the open during the Phase II project. There are specific areas we are addressing in this work, but the intent is to create a general purpose, extensible framework that will be useful across disciplines from quantum chemistry, through to molecular dynamics, materials science, and biology. I think it is only by developing open, cross-platform solutions that are well tested using the Kitware software process that we will truly be able to innovate in chemistry. We hope to continue engaging in collaborations with other leaders in the field to produce the best solutions, and to help further the goals of open chemistry, and open science in general, by making it easier to share data and collaborate.
References
[1] http://hutchison.chem.pitt.edu/
[2] http://avogadro.openmolecules.net/
[3] http://openbabel.org/
[4] http://www.msg.ameslab.gov/gamess/
[5] http://blueobelisk.sourceforge.net/
[6] http://cactus.nci.nih.gov/chemical/structure
[7] http://quixote.wikispot.org/
[8] http://www.mongodb.org/
 Marcus Hanwell is an R&D Engineer in the scientific computing team at Kitware. He has a background in open source, open science, Physics and Chemistry. He developed large portions of the new charting framework in VTK, and also contributes to ParaView, Titan, ITK, CMake and software quality process.
Marcus Hanwell is an R&D Engineer in the scientific computing team at Kitware. He has a background in open source, open science, Physics and Chemistry. He developed large portions of the new charting framework in VTK, and also contributes to ParaView, Titan, ITK, CMake and software quality process.